 0800 2863 2863
0800 2863 2863 info@bv-nf.de
info@bv-nf.deGenetik der NF2
Die NF2 gehört zu den so genannten monogenen Erkrankungen, bei denen Veränderungen (Mutation) in nur einem Gen die Störung bewirken. Jeder, der eine solche Mutation trägt, zeigt auch klinische Anzeichen der Erkrankung, allerdings in sehr unterschiedlichem Grad der Ausprägung.
Ursache für die NF2 ist die Mutation eines Gens auf dem Chromosom 22, das vermutlich Einfluss auf Form und Wanderungsverhalten bestimmter Zelltypen nimmt. Das Produkt, dessen Entstehung dieses NF2-Gen steuert, ist eine Kette von 595 Aminosäuren (also ein Eiweiß) und wird Merlin oder Schwannomin genannt.
Mutationen im Schwannomin-Gen oder das Fehlen von Schwannomin führen dazu, dass die Zellwanderung beeinflusst wird, die Zellform sich ändert oder ein Verlust der sog. Zellkontakthemmung eintritt.

(Pete Linforth auf Pixabay)

Genetische Grundlagen zur NF2
Mutationen selbst sind nichts Ungewöhnliches und gehören zu unserer Existenz. Ohne Mutationen gäbe es keine Evolution, also auch nicht uns Menschen. Mutationen sind sozusagen der Motor der Evolution. Sie machen das aus, was wir Individualität nennen.
Auch Menschen, die nicht an erblichen Erkrankungen leiden, weisen in ihren Körperzellen viele Mutationen auf, z.B. in Hautzellen nach einem Sonnenbad. Man nennt diese Art von Mutationen „somatische“ Mutationen. Die meisten von ihnen werden von den Körperzellen wieder repariert, so z.B. diejenigen, die durch Sonnenbestrahlung bewirkt werden.
Durch die notwendigen Reparaturmechanismen nicht mehr reparierbare Mutationen sind für die Zellen oft bedeutungslos, gelegentlich aber auch die Ursache für die Umwandlung der betroffenen Zelle in eine Tumorzelle oder sie verursachen Erkrankungen wie Hypercholesterinämie, Gicht oder bestimmte neurologische Erkrankungen.

DNA
Eine Mutation entsteht folgendermaßen: Träger der genetischen Information ist die DNA (Desoxyribonukleinsäure, desoxyribonuclein acid). Die DNA-Moleküle in jeder menschlichen Zelle ergäben aneinander gereiht einen bis zu zwei Meter langen Faden. Daher sind diese Molekülketten in der Zelle dicht geknäuelt gelagert. Sie sind in 46 Abschnitte unterteilt, so genannte Chromosomen.
Die DNA-Moleküle werden nur aus jeweils vier Bausteinen gebildet. Es sind dies die 4 Basen Adenin (A), Guanin (G), Cytosin (C) und Thymin (T). Die Abfolge dieser Bausteine legt das genetische Programm der Zelle fest. Genauer betrachtet besteht die DNA aus zwei Molekülsträngen, die sich wie ein Reißverschluss zusammenlagern. Dabei bilden die gegenüberliegenden Basen (Nukleobasen) feste Paare: A mit T und G mit C.
Jeweils drei Basenpaare bestimmen (kodieren) dabei den Einbau einer bestimmten Aminosäure in ein Protein. Die Gene enthalten somit die Information darüber, in welcher Reihenfolge die Aminosäuren miteinander verknüpft werden sollen.
Jede Zelle eines Menschen besitzt nun zwei Kopien jedes Gens. Eine davon wurde von der Mutter geerbt, die andere vom Vater. Bei NF2 Betroffenen befindet sich auf mindestens einem dieser beiden Gene eine Veränderung, die die Funktion dieses Gens stört.
Die eigentliche Tumorentstehung kommt allerdings dadurch zustande, dass zufallsmäßig das zweite NF2 Gen auch eine Mutation erfährt. Man spricht hier von einer zusätzlichen somatischen Mutation, von einem "second hit".
Somatische Mutationen im NF2 Gen sind allerdings nichts Ungewöhnliches, und sogar nicht von der NF2 Betroffene weisen oft in diesem NF2 Gen Mutationen auf. Bei NF2 Betroffenen führt die Mutation im zweiten NF2 Gen allerdings zum vollständigen funktionellen Verlust des zugehörigen Proteins.
In den von der Zweitmutation betroffenen Zellen gibt es also kein funktionsfähiges Schwannomin/Merlin mehr. Dies führt dazu, dass die das Wachstum bremsende Funktion entfällt und die Zellen sich unkontrolliert weiter teilen. Aus diesen Zellen entstehen dann die Tumoren.
In den Schwannomen, den häufigsten Tumoren der NF2, findet sich kein gesundes (unverändertes) NF2-Gen mehr. Unklar ist dagegen noch die Beteiligung des NF2-Gens z.B. an der Bildung von Meningeomen, d.h. Wucherungen der Hirnhäute. Hier wird angenommen, dass für die Entstehung dieser bei NF2 häufigen Tumoren möglicherweise noch Veränderungen in anderen Genen vorliegen.
Funktionell können NF2 Mutationen dazu führen, dass eines der beiden NF2 Gene
- komplett seine Funktion verliert (loss of function mutation, Null Mutationen)
- für ein nur noch unzureichend seine normale Funktion ausführendes Protein codiert
- ein Protein kodiert, das nicht nur seine normale Funktion nicht mehr ausüben kann, sondern zusätzlich noch eine weitere Funktion aufweist (gain of function mutation)
Man unterscheidet dementsprechend:
- Spleißmutation:
Die Gene höherer Organismen, also auch die des Menschen, besitzen innerhalb des Genbereiches nicht-codierende Bereiche. Nach der Umschreibung der DNA in RNA werden diese herausgeschnitten, so dass die codierenden Abschnitte zur vollständigen mRNA zusammengespleißt werden können. Damit dieser Spleißprozess ordnungsgemäß ablaufen kann, müssen bestimmte Erkennungssequenzen vorhanden sein, die den Spleißapparat an die richtige Stelle führen. Auch in diesen Erkennungssequenzen sind Mutationen möglich, die zu verschiedenartigen Störungen des Spleißprozesses führen. Entsprechend kommen dann natürlich auch unterschiedliche Formen des codierten Proteins vor. - Nonsense Mutationen:
Diese Mutationen gehen mit Leserasterveränderungen einher. Dies führt dazu, dass es beim Umsetzen der Information zu einem vorzeitigen Stopsignal kommt. Das gebildete Protein wird also kürzer als das eigentliche Schwannomin. - Missense Mutationen:
Missense Mutationen führen zum Austausch einer Aminosäure gegen eine andere. Solche Mutationen sind für die Funktionsanalyse von Proteinen besonders wertvoll, da sie unmittelbare Rückschlüsse auf die Bedeutung der jeweils ausgetauschten Aminosäure erlauben. Eine Reihe der Missense Mutationen beeinträchtigen die Funktion des NF2-Gens Schwannomin, heben sie aber nicht auf. Missense Mutationen lassen die Bildung eines annähernd normalen Proteinproduktes zu.
Neuere Befunde wissenschaftlicher Studien sprechen dafür, dass für die Ausprägung des Krankheitsbildes der NF2 auch noch andere Gene oder Umweltfaktoren verantwortlich sind. Für diese Tatsache spricht z.B., dass es zwar klinisch gesicherte Fälle von NF2 gibt, bei denen aber die Mutation des NF2 Gens nicht genau lokalisiert werden konnte. Auch gibt es Fälle, in denen Patienten mit der gleichen Mutation unterschiedliche Verlaufsformen zeigen.
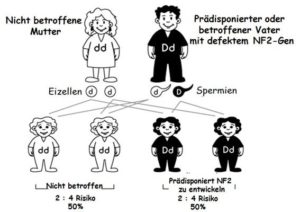
Das Risiko, die Erkrankung zu vererben, beträgt statistisch gesehen zunächst 50%, sofern die Mutation auch in den Keimzellen vorliegt (dies ist nicht zwingend der Fall).
Tritt die NF2 erstmalig in einer Familie auf, spricht man von einer Neumutation. Ungefähr die Hälfte aller NF2-Patienten sind eine solche Spontan- oder Neumutation. Diese Patienten haben also ihre Mutation nicht von einem der beiden Elternteile, die demnach auch keine Symptome der Krankheit zeigen, geerbt.
Nur wenn die Mutation auch innerhalb einer elterlichen Keimzelle (Ei- oder Samenzelle) besteht, weist das Kind die genetische Veränderung in all seinen Körperzellen auf. Dies ist besonders folgenhaft, weil sich aus einer Keimzelle bis zur Geburt ca. 70 000 000 000 000 weitere Zellen entwickeln. Körperzellen, die eine Mutation erfahren, teilen sich in der Regel aber seltener.
Bei ca. 30% aller NF2-Patienten mit einer Neumutation ist die NF2-Mutation aber nicht in einer der Keimzellen aufgetreten, sondern in einer Köperzelle eines erst späteren embryonalen Entwicklungsstadiums. Diese Patienten sind so genannte Mosaikfälle. Sie können anschließend die Krankheit auf ihre Nachkommen vererben (wenn auch ihre Fortpflanzungszellen (Ei- oder Samenzellen) von der Mutation betroffen sind), welche dann auch eine stärkere Ausprägung der Krankheit mit früherem Auftreten der Symptome zeigen können als ihre Eltern.
Einen Mosaikfall kann man sich folgendermaßen vorstellen: Jede Körperzelle trägt je eine Kopie der Erbinformation, wie sie ursprünglich in Ei- oder Samenzelle vorhanden war. Tritt nun erst nach den ersten Zellteilungen während der Embryonalentwicklung eine Mutation im NF2 Gen auf, so sind nicht alle Zellen des sich entwickelnden Menschen von dieser Mutation betroffen. Man spricht dann von einem genetischen Mosaik für eine Mutation im NF2 Gen.
Bei den Mosaikfällen lässt sich zwischen somatischen Mosaiken und Keimzellmosaiken unterscheiden.
Bei somatischen Mosaiken trägt ein Teil der Körperzellen die Mutation.
Keimzellmosaik heißt dagegen, dass die Mutation sich auch auf einen Teil der Keimzellen bezieht.
Klinisch kann man nicht zwischen somatischen Mosaiken und Keimzellmosaiken unterscheiden. Eine solche Unterscheidung wäre aber wünschenswert, da das Risiko, die NF2-Mutation zu vererben erhöht ist, wenn die Keimzellen mit betroffen sind. Genetische Untersuchungen der Keimzellen können hier weiterhelfen.
Das Risiko der Vererbung der NF2 kann bei Mosaiken geringer sein als bei Betroffenen, die in all ihren Zellen die Mutation aufweisen. Theoretisch kann ein sporadischer NF2-Betroffener viele Symptome der NF2 zeigen, aber gleichzeitig die Erkrankung nicht an seine Kinder weitervererben, weil die Mutation die Keimzellen nicht betrifft.
Man kann allerdings nur mit sehr aufwändigen Methoden feststellen, ob bei einem Betroffenen auch die Keimzellen verändert sind. Die Untersuchung der Samenzellen der Männer ist dabei technisch leichter und komplikationsloser durchzuführen als die Untersuchung der Eizellen einer Frau. Bei Entnahme von Eizellen aus diagnostischen Gründen lässt sich auch nur ein statistisches Risiko ermitteln, da die untersuchten Keimzellen selbst ja für die Befruchtung verloren sind. Es lässt sich daher in der genetischen Beratung nicht exakt beziffern, wie hoch das Risiko ist, die NF2 weiterzuvererben.
Bei einem Mosaikfall ist ebenfalls die molekulargenetische Untersuchung erschwert. Da die Mutation nur in einem Teil der Körperzellen vorhanden ist, kann sie häufig in den Blutzellen des Patienten nicht nachgewiesen werden. Untersuchungen zum Nachweis der Mutation in Tumorgewebe können allerdings in solchen Fällen die diagnostische Treffsicherheit erhöhen.
In Studien, die die Ausprägung der Erkrankung, ihren Verlauf und den Typ der Mutation genau untersuchen, versucht man die Frage zu beantworten, ob bestimmte Mutationstypen mit bestimmten Krankheitserscheinungen eng verbunden sind. Man möchte am liebsten schon bei einem noch klinisch unauffälligen Patienten mit großer Sicherheit den Krankheitsverlauf vorhersagen können, um die Therapie zu optimieren.